The Truth About Bioavailability of Colloidal Silver
Much has been written concerning the bioavailability of colloidal silver products, most of which misrepresents the true meaning of the term.
From reading the section below from the Merck Manual it is clear that bioavailability is the amount of unchanged drug that reaches the systemic circulation. In that regard, it pertains to how colloidal silver particles are absorbed into the bloodstream after being ingested. We know that silver nanoparticles are absorbed in the first few feet of the small intestine and their presence can be measured using atomic absorption/emission spectroscopy beginning about 15 minutes after ingestion. The silver concentration of nanoparticles in the blood serum will continue to increase for about 3 to 4 hours after ingestion and then start to taper off in accordance with the graph (Fig.1). True silver colloids reach the bloodstream unchanged in form, meaning they are truly bioavailable. Ionic silver does not reach the bloodstream unchanged because the silver ions are converted into silver chloride which means ionic silver products are less bioavailable than true colloidal silver products.
Silver is a noble metal and in its elemental state (metallic, not ionic) does not readily combine to form compounds. The biological activity of metallic silver nanoparticles does not require that the silver combines with biological material in the body. Its action is one of being a catalyst, and as such, it remains unchanged from its original form (silver nanoparticles).
Silver nanoparticles, which are the predominant form of silver in true colloidal silver products, do not dissolve nor are they metabolized in the human body. They enter the body as silver nanoparticles, they circulate in the bloodstream and then leave the body and always remain as silver nanoparticles. They perform their catalytic action while in the form of silver nanoparticles; their effectiveness is determined by their particle surface area. For more details, see Particle Surface Area and Effectiveness.
Notice that the definition of bioavailability only concerns the amount of drug (or other substance) that reaches the systemic circulation, it does not have anything to do with the ability of the substance to be metabolized by the body. It does not have anything to do with the substance being water-soluble or combining with biological tissue. These are important to remember because so much has been written that claims that silver nanoparticles are not bioavailable when clearly any such statement is false. If they were not bioavailable, they would not enter the bloodstream.
The section below is excerpted from the Merck Manual of Diagnosis and Therapy, Section 22 Clinical Pharmacology.
bioavailability – Extent to which–and sometimes rate at which–the active moiety (drug or metabolite) enters systemic circulation, thereby gaining access to the site of action. The physicochemical properties of a drug govern its absorptive potential, but the properties of the dosage form (which partly depend on its design and manufacture) can largely determine drug bioavailability.
Differences in bioavailability among formulations of a given drug can have clinical significance. Thus, the concept of equivalence among drug products is important in making clinical decisions. Chemical equivalence refers to drug products that contain the same compound in the same amount and that meet current official standards; however, inactive ingredients in drug products may differ. Bioequivalence refers to chemical equivalents that, when administered to the same person in the same dosage regimen, result in equivalent concentrations of drug in blood and tissues. Therapeutic equivalence refers to drug products that, when administered to the same person in the same dosage regimen, provide essentially the same therapeutic effect or toxicity. Bioequivalent products are expected to be therapeutically equivalent. Therapeutic problems (eg, toxicity, lack of efficacy) are encountered most frequently during long-term therapy when a patient who is stabilized on one formulation is given a nonequivalent substitute (as for digoxin or phenytoin). Sometimes therapeutic equivalence may be achieved despite differences in bioavailability. For example, the therapeutic index (ratio of the maximum tolerated dose to the minimum effective dose) of penicillin is so wide that moderate blood concentration differences due to bioavailability differences in penicillin products may not affect therapeutic efficacy or safety. In contrast, bioavailability differences are important for a drug with a relatively narrow therapeutic index. The physiologic characteristics and comorbidities of the patient also affect bioavailability. The absorption rate is important because even when a drug is absorbed completely, it may be absorbed too slowly to produce a therapeutic blood level quickly enough or so rapidly that toxicity results from high drug concentrations after each dose.

Causes of Low Bioavailability
When a drug rapidly dissolves and readily crosses membranes, absorption tends to be complete, but the absorption of orally administered drugs is not always complete. Before reaching the vena cava, a drug must move down the GI tract and pass through the gut wall and liver, common sites of drug metabolism; thus, a drug may be metabolized (first-pass metabolism) before it can be measured in the systemic circulation. Many drugs have low oral bioavailability because of extensive first-pass metabolism. For such drugs (eg, isoproterenol, norepinephrine, testosterone), extraction in these tissues is so extensive that bioavailability is virtually zero. For drugs with an active metabolite, the therapeutic consequence of the first-pass metabolism depends on the contributions of the drug and the metabolite to the desired and undesired effects. Low bioavailability is most common with oral dosage forms of poorly water-soluble, slowly absorbed drugs. More factors can affect bioavailability when absorption is slow or incomplete than when it is rapid and complete, so slow or incomplete absorption often leads to variable therapeutic responses. Insufficient time in the GI tract is a common cause of low bioavailability. The ingested drug is exposed to the entire GI tract for no more than 1 to 2 days and to the small intestine for only 2 to 4 h. If the drug does not dissolve readily or cannot penetrate the epithelial membrane (eg, if it is highly ionized and polar), time at the absorption site may be insufficient. In such cases, bioavailability tends to be highly variable as well as low. Age, sex, activity, genetic phenotype, stress, disease (eg, achlorhydria, malabsorption syndromes), or previous GI surgery can affect drug bioavailability. Reactions that compete with absorption can reduce bioavailability. They include complex formation (eg, between tetracycline and polyvalent metal ions), hydrolysis by gastric acid or digestive enzymes (eg, penicillin and chloramphenicol palmitate hydrolysis), conjugation in the gut wall (eg, sulfoconjugation of isoproterenol), adsorption to other drugs (eg, digoxin and cholestyramine), and metabolism by luminal microflora.
Assessment of Bioavailability
Assessment of bioavailability from plasma concentration-time data usually involves determining the maximum (peak) plasma drug concentration, the time at which maximum plasma drug concentration occurs (peak time), and the area under the plasma concentration-time curve (AUC) as indicated by Figure 1 below.
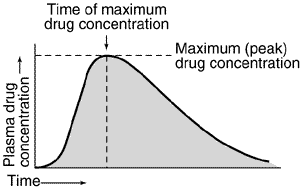
Fig.1 – Representative plasma concentration-time relationship after a single oral dose of a hypothetical drug. The area under the plasma concentration-time curve is indicated by shading.
The plasma drug concentration increases with the extent of absorption; the peak is reached when the drug elimination rate equals the absorption rate. Bioavailability determinations based on the peak plasma concentration can be misleading because drug elimination begins as soon as the drug enters the bloodstream. The most widely used general index of the absorption rate is peak time; the slower the absorption, the later the peak time. However, peak time is often not a good statistical measure because it is a discrete value that depends on the frequency of blood sampling and, in the case of relatively flat concentrations near the peak, on assay reproducibility.
AUC is the most reliable measure of bioavailability. It is directly proportional to the total amount of unchanged drug that reaches the systemic circulation. For an accurate measurement, blood must be sampled frequently over a long enough time to observe virtually complete drug elimination. Drug products may be considered bioequivalent in extent and rate of absorption if their plasma-level curves are essentially superimposable. Drug products that have similar AUCs but differently shaped plasma-level curves are equivalent in extent but differ in their absorption rate-time profiles.
Related Information
- How to Compare Colloidal Silver Products
- The Truth About Ionic Silver
- The Truth About Covalent Silver
- Myth of Monatomic Colloidal Silver
- Particle Surface Area and Effectiveness
- The Truth About TEM Images of Ionic Silver Solutions
- The Truth About Silver Protein Products
- Bogus Scientific Claims Made for Colloidal Silver Products
- The Truth About Colloid Particle Size
- Find colloidal silver products on the internet